摘要
本文研究了使用高比例水相流动相(水比例 > 90%)时,C₈和C₁₈高效液相色谱柱出现保留下降的原因。我们描述了一种用于定量该保留下降程度的方法,该方法涉及停泵后重新启动流动。
利用这一方法,我们研究了保留下降与键合相孔径、表面浓度及化学结构的关系。
此外,我们还通过改变流动相组成或提高柱压,探索了如何恢复色谱柱原有的保留能力。结果表明,这些现象与基于“非润湿液体填充孔隙”理论的机理一致,该理论也用于压汞法孔隙率测定。
我们认为,保留下降的原因是:当流动相停止、压力释放后,高比例水相被迫从孔隙中排出。由于流动相不再与颗粒内部表面(即大部分表面积所在之处)接触,因此保留能力下降。
最后,本文还讨论了该机理对最大化极性分析物反相保留效果的启示意义。
关键词: 反相;HPLC;高水相流动相;保留下降
引言
在反相液相色谱中,为了保留极性较强的分析物(否则它们会在死时间出峰),通常需要使用高比例水相的流动相。
然而,许多研究发现:当水相比例超过90%时,使用C₈或C₁₈色谱柱会出现一些异常现象。这些现象通常解释为:在高比例水相下,键合在硅胶表面的烷基链发生聚集,导致分析物无法接触到它们,从而引起保留下降。
1997年,我们针对“停泵后重新启动时保留下降”这一现象提出了另一种解释——流动相被从颗粒的孔隙中“挤”了出来。该观点基于两个关键观察:保留下降程度与键合相的孔径有关,且柱压是一个重要变量。
我们曾部分报道过这些结果。相同的理论也被用来解释反相固相萃取中吸附剂床层在活化后变干所导致的保留下降。
此后,其他研究者也发表了类似的观察结果,但提出了不同的解释。例如,Reid 和 Henry 将保留下降归因于固定相烷基链的折叠;Bidlingmeyer 和 Broske 则认为,“主要驱动力是高比例水相在固定相表面形成界面结构”。
在本报告中,我们首先介绍了一种用于测定停泵并重新启动后保留下降程度的方法(使用高比例水相)。利用该方法,我们研究了保留下降与孔径、键合相表面浓度及化学结构的关系。
我们还报道了两种恢复保留的方法:提高流动相中有机相的比例,以及提高色谱柱的柱压。实验结果强烈支持“流动相被挤出孔隙”这一机制。最后,我们讨论了该机制对最大化极性分析物反相保留效果的启示。
实验部分
仪器设备
元素分析使用 240XA CHN 分析仪完成。比表面积(As)、比孔容(Vp)和平均孔径(Dp)采用多点氮气吸附法测定,使用 ASAP 2400 型仪器。
As值通过多点 BET 法计算,Vp值在 P/P0>0.98 的单点条件下测定,Dp值由吸附等温线的脱附支通过 BJH 法计算。键合相的表面浓度使用 Berendsen 和 de Galan 的方程计算。
HPLC 分析条件
使用 Waters 公司的 ExpertEase V3.2 色谱管理软件进行仪器控制、数据采集和处理。
色谱系统由以下 Waters 组件构成:一台 600 型溶剂输送系统、一台 490E 型可编程波长紫外检测器(检测波长设为 254 nm)以及一台 717plus 型自动进样器。
柱温由 Euramark生产的 Mistral 型恒温柱箱控制,设定为 25°C。
除高压复湿实验外,整个测试过程中流速均设为 1 mL/min。
样品为溶于 80/20(v/v)D₂O/甲醇中的 100 μg/mL 磺胺和 100 μg/mL 普鲁卡因胺,进样量为 10 μL。
保留因子根据三次重复进样的平均保留时间计算,以 D₂O 作为死体积标记物。流动相为 20 mM K₂HPO₄(pH 6.00)水溶液,或分别加入 5% 或 10%(v/v)甲醇。
测定保留下降的色谱流程
色谱柱首先在 1 mL/min 流速下用 100% 甲醇平衡 30 min,再用 50/50 甲醇/水平衡 30 min,分别用于确保固定相完全润湿以及防止磷酸盐缓冲液(pH = 6)析出。
然后,在磷酸盐流动相(pH = 6)中平衡 30 min,之后进行三次进样。运行时间设置足够长以保证所有组分洗脱完全:对于含 0%、5% 和 10% 甲醇的流动相,典型运行时间分别为 60 min、20–25 min 和 10 min。进行三次重复进样以测定保留时间,取平均值计算保留因子。
完成初始进样后,在 4 min 内将流速降至 0,并保持 0 流速 1 小时(除非另有说明)。流速停止结束后,在 15 秒内将流速恢复至 1 mL/min。约 1 分钟后,再进行三次重复进样测定保留时间,取平均值计算保留因子。
上述流程首先使用 100% K₂HPO₄ 流动相(ph = 6)进行,然后依次使用含 5% 和 10% 甲醇的流动相进行。保留下降百分比的计算方式为:用停流前后每种测试物质的保留因子差值,除以初始保留因子,再乘以100%。
恢复保留的色谱流程
为考察通过压力恢复保留的效果,在停泵后通过逐步提高流速来增加柱压。采用上述保留下降流程,但仅使用 100% 水相流动相,停泵时间设为 10 min(已确定该时间足以使本研究所用键合相发生完全保留下降)。
在停泵并以 1 mL/min 重新启动后测定保留下降,随后将流速升至 1.5 mL/min 并保持 30 min,然后再次进行三次重复进样重新评估保留下降。依次在 2.0、2.5 和 3.0 mL/min 流速下重复该流程。
采用类似方法确定在 100% 水相流动相中停流后,恢复 C₁₈ A 键合相保留所需的最低甲醇百分比。
在停泵并以 1 mL/min 重新启动后测定保留下降,然后将色谱柱在 1 mL/min 下用 10/90 甲醇/水(v/v)平衡 30 min,再重新用 100% 水相流动相(pH = 6)平衡 30 min,之后进行三次重复进样重新评估保留下降。依次使用 20/80、30/70、40/60 和 50/50(v/v)甲醇/水流动相重复该流程,每次之后再用 100% 水相流动相重新评估保留下降。
试剂与材料
用于许多样品的孔径为 9.2 nm 的硅胶为 5.0 μm Symmetry 硅胶。更大孔径的硅胶样品通过对该硅胶进行水热扩孔处理获得。
所有键合相均使用氯代二甲基硅烷制备,并用三甲基硅基进行封端。高表面浓度键合相使用相对于硅胶反应量过量的硅烷制备。降低表面浓度的 C₁₈ 材料使用化学计量比的N-十八烷基二甲基氯硅烷制备。
N-十八烷基氨基甲酸酯键合相(C₁₈-氨基甲酸酯)的结构与制备方法已有报道。本研究所用材料的碳含量、表面浓度、比表面积、孔容和平均孔径汇总于表 1(表见英文原文)。
所有材料均采用专有的高压匀浆装填法装入 150 mm × 3.9 mm 不锈钢色谱柱中。所有试剂和溶剂均直接使用,未进一步纯化。
结果
保留下降的定量方法
有报道指出,当某些反相色谱柱使用高比例水相时,保留时间会逐渐提前。然而,我们在使用高表面浓度的C₁₈键合相柱时,在连续运行20小时、流动相为纯水相的条件下,并未观察到保留变化。
但是,当停泵后再重新启动时,我们看到保留能力几乎完全丧失。当使用甲醇或乙腈体积分数超过10%的流动相时,则未观察到这种保留下降。
基于这一观察,我们设计了一个流程来定量测定使用高比例水相时因停泵再启动所导致的保留下降。
先将色谱柱(150 mm × 3.9 mm)用100%甲醇冲洗30 min,再用50/50(v/v)甲醇/水以1 mL/min冲洗30 min,然后用高比例水相流动相平衡30 min。进样测试混合物,包含磺胺、普鲁卡因胺以及作为死体积标记物的D₂O。
待所有分析物洗脱完毕后,停泵预定时间,然后以1 mL/min重新启动。保留下降程度:用停泵前后普鲁卡因胺的保留因子差值占初始保留因子的百分比表示。实验误差范围内,普鲁卡因胺和磺胺的相对保留下降程度相同。
我们通过改变停泵时间研究了保留下降的动力学。对于高表面浓度键合相,停泵10分钟后保留下降即达到恒定值。然而,对于表面浓度为2.00 μmol/m²的C₁₈键合相(C₁₈ E),保留因子在停泵9小时后仍持续下降。
在后续实验中,除非另有说明,停泵时间均采用1小时。除了目标分析物保留下降外,我们还观察到死体积标记物D₂O的洗脱时间也减少了。对于装有C₁₈ A键合相的150 mm×3.9 mm色谱柱,死体积从1.24 mL降至0.95 mL。这0.29 mL的体积变化与柱内颗粒孔隙中所含流动相的体积(1.0 g/柱 × 0.31 mL/g = 0.31 mL/柱)相近。
对于表面浓度为2.00 μmol/m²的C₁₈ E键合相柱,死体积变化较小(0.08 mL),该材料在停流后的保留下降也较小(1小时后为18%,见下文)。McCormick 和 Karger 在使用纯水流动相与C₈柱时也报道了类似的D₂O洗脱时间变化。
3.2 保留下降对孔径的依赖性
为了研究保留下降对填料孔径的依赖性,我们测试了一系列高表面浓度、封端的C₁₈键合相,这些材料采用平均孔径从9.3 nm到24.5 nm的硅胶制备。键合后的平均孔径在6.7 nm到19.9 nm之间。孔径减小是已知现象,由键合基团部分填充孔网络所致。
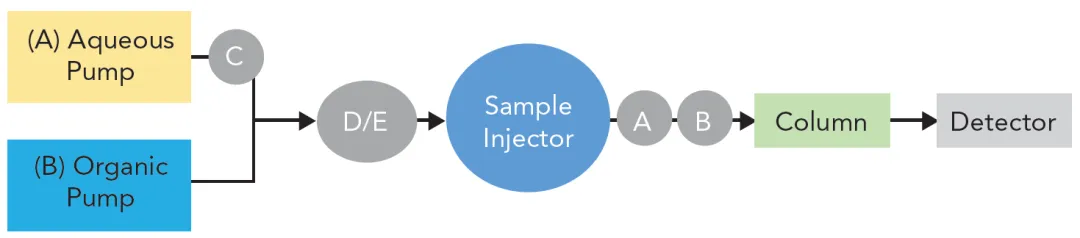
图1:对于高表面浓度C₁₈键合相,停泵再启动后普鲁卡因胺的保留下降与平均孔径的关系。方块:100% 20 mM K₂HPO₄ pH 6流动相;三角:95/5缓冲液/甲醇;菱形:90/10缓冲液/甲醇。
图1显示了在三种不同流动相组成下,停泵再启动后保留下降对键合相孔径的依赖关系。使用100%水相流动相时保留下降最大。对于最小孔径的材料,在该流动相下保留下降接近100%;而对于最大孔径的材料,保留下降仅为8%。其他研究者也报道过这种对孔径的依赖性。
保留下降对C₁₈表面浓度的依赖性
为了研究保留下降对表面浓度的依赖性,我们测试了一系列在孔径约9.1–9.3 nm硅胶上制备的C₁₈键合相。五种材料的C₁₈表面浓度在2.00到3.21 μmol/m²之间(表1中C₁₈ A和E–H),且均经过完全封端。
图2显示了在三种不同流动相组成下,保留下降对C₁₈表面浓度的依赖关系。同样,使用100%水相流动相时保留下降最大。在该流动相下,两者呈现出很强的相关性:保留下降随C₁₈表面浓度降低而减小。Bidlingmeyer 和 Broske 也报道了类似的趋势。
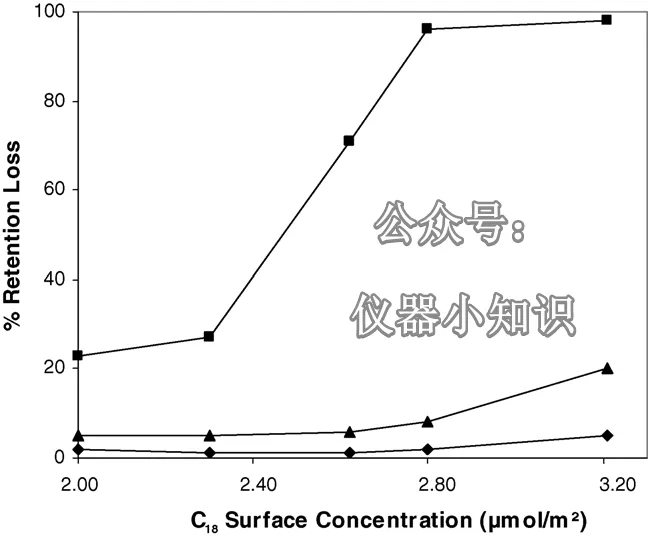
图2:对于基于平均孔径9.1–9.3 nm硅胶的C₁₈键合相,停泵再启动后普鲁卡因胺的保留下降与C₁₈表面浓度的关系。方块:100% 20 mM K₂HPO₄(pH = 6)流动相;三角:95/5缓冲液/甲醇;菱形:90/10缓冲液/甲醇。
保留下降对键合相化学结构的依赖性
为了考察保留下降如何随键合相的化学结构变化,我们测试了三种高表面浓度、封端的材料:C₁₈ A、C₈ 以及 N-十八烷基氨基甲酸酯(C₁₈-氨基甲酸酯)。这些材料的特性列于表1。
同样,使用100%水相流动相时保留下降最大。在该流动相下,C₁₈ A 和 C₈ 键合相均表现出98%的保留下降,而 C₁₈-氨基甲酸酯键合相的保留下降不到3%。C₁₈ A 和 C₈ 键合相在使用95/5缓冲液/甲醇流动相时也显示出显著的保留下降,其中C₈材料的保留下降更大(84%),而C₁₈ A材料为20%。
恢复保留
当因停泵导致保留下降后,可以通过用较高甲醇浓度的流动相重新平衡色谱柱来恢复原有的保留。为了确定恢复原有保留所需的甲醇浓度,我们使用装有高表面浓度、孔径9.3 nm的C₁₈ A键合相的色谱柱进行了实验。
按照上述保留下降流程,使用100%水相流动相并停泵10 min后,色谱柱几乎完全丧失保留(下降接近100%)。
然后,依次用不同比例的甲醇/水流动相冲洗色谱柱,从10%甲醇开始,逐渐增加到50%。每种流动相均以1 mL/min通过色谱柱30 min,随后用100%水相流动相以1 mL/min平衡30 min,再进样测定目标分析物的保留因子。
结果如图3所示。随着再生流动相中甲醇含量的增加,保留逐渐恢复到原始值。对于这种孔径9.3 nm、高表面浓度的C₁₈键合相,需要40%的甲醇才能恢复原始保留。其他研究者也报道过,使用约50%甲醇或乙腈的流动相可以恢复保留。
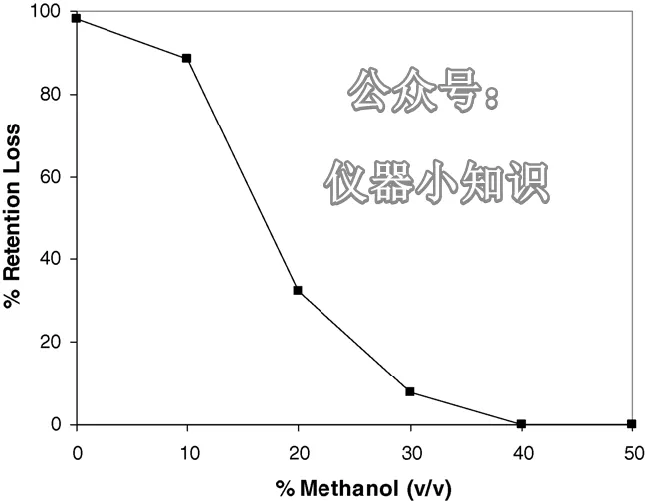
图3:对于C₁₈ A键合相,停泵再启动后普鲁卡因胺的保留下降与再生流动相中甲醇含量的关系。
另一种恢复保留的方法是保持100%水相流动相,通过提高流速来增加柱压。为了研究压力对保留的影响,我们再次使用装有高表面浓度、孔径9.3 nm的C₁₈ A键合相的色谱柱。在按照标准保留下降流程(100%水相流动相,停泵10 min)后,色谱柱几乎完全丧失保留。
然后,在同样使用100%水相流动相的条件下逐步提高流速。记录每个流速下的柱压,并进样测定目标分析物的保留因子。结果如图4所示。当压力升高至22.8 MPa(228 bar,3300 psi)时,保留有所恢复。然而,即使在34.5 MPa的压力下,也只能恢复到原始保留的62%。当流速降回1 mL/min(此时柱压为11.0 MPa)时,保留保持在与升压后相同的水平。之所以无法通过这种方式完全恢复保留,是因为色谱柱出口端仍处于大气压。
为了完全恢复原始保留,需要在色谱柱后加装限流装置,使柱出口端保持高于大气压。其他研究者也报道过压力至少能部分恢复保留。但 Reid 和 Henry 指出,对于他们研究的色谱柱,这种方法只有在加压时柱内含有一些有机溶剂(如10%乙腈)的情况下才有效。
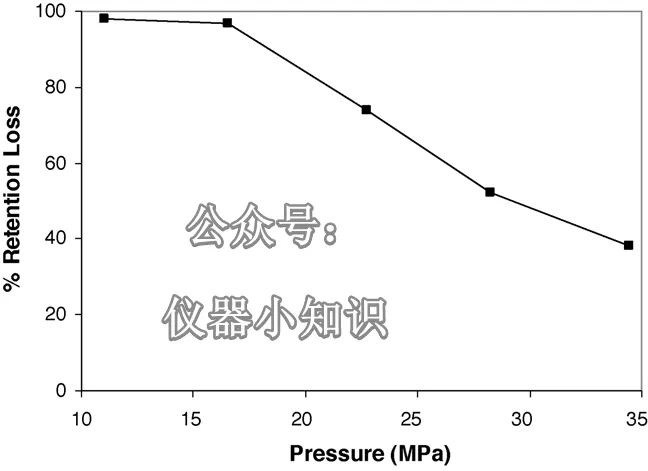
图4:对于C₁₈ A键合相,使用100% 20 mM K₂HPO₄(pH = 6)流动相时,普鲁卡因胺的保留下降与压力的关系。
讨论
我们认为,最能解释上述观察结果的机制是基于“非润湿液体填充孔隙”的理论。这也是压汞法测定多孔固体孔径分布的常用原理。
在压汞实验中,先将多孔固体样品抽真空,然后在压力下将汞压入孔隙中。汞对许多材料是不润湿的。压入样品中的汞体积作为施加压力的函数被测量出来。将汞压入半径为r的圆柱形孔隙所需的压力(ΔP)由 Washburn 方程给出:
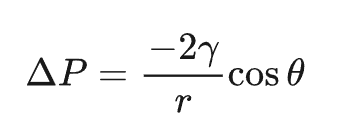
其中γ 是汞的表面张力,θ是汞与样品之间的接触角。利用该方程,可以将压入体积与压力的关系数据转换为孔体积与孔径的关系图。
上述结果可以用类似的方式解释。已知大多数 C₁₈ 键合相不被水润湿。根据定义,这意味着 θ> 90°。
尽管测定非润湿液体接触角的常规方法不适用于典型的色谱颗粒,但 Engelhardt 和 Mathes 描述了一个简单的测试键合相润湿性的方法:将键合相样品与水一起振荡,观察样品是浮在水面上(不润湿)还是下沉(润湿)。
通过该测试,本工作中使用的所有材料均不被纯水润湿。为了定量估计水在 C₁₈ 硅胶上接触角大于 90°时的数值,我们需要参考在平面表面上测得的数值。
Montgomery 等人报道,在键合了十八烷基二甲基氯硅烷并用三甲基氯硅烷封端的硅片上,纯水的接触角为 93°。Maoz 和 Sagiv 报道,在玻璃片上自组装的十八烷基三氯硅烷单分子层上,纯水的前进接触角为 112°。
Wasserman 等人报道,在硅片表面的烷基三氯硅烷(丁基及更长链)形成的薄膜上,纯水的前进接触角为 110°,并指出这些薄膜的后退接触角比前进接触角低 10°。
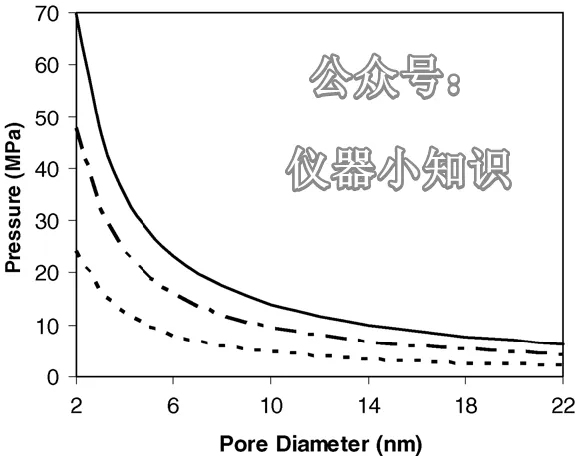
图5. 将纯水强行注入不同直径的孔隙时所需要计算出的压力:(—) θ = 120°、(— – —) θ = 110°,(- – -) θ = 100°。
另一个有用的参考值是 Janczuk 等人测量的纯水在石蜡薄膜上的接触角为 110.6°。Janczuk 等人还报道了不同比例甲醇、乙醇和丙醇与水的混合物在石蜡薄膜上的接触角。他们发现,润湿石蜡所需的甲醇浓度介于 20% 到 40% 之间。
由于纯水对我们所研究的材料是不润湿的,因此除非施加压力,否则水不会进入孔隙。
对于不同的孔径和接触角,可以使用 Washburn 方程计算将水压入孔隙所需的压力,其中 γ 取水的表面张力(72 dyn/cm)。(我们忽略了 20 mM 磷酸盐缓冲液引起的表面张力微小增加)
但需要注意的是,像乙酸这样的有机添加剂会显著降低表面张力。)对于 θ = 120°、110° 和 100° 的计算结果如图 5 所示。
当压力低于这些计算值时,水会被迫从孔隙中排出。以 θ = 110° 为例,将水压入孔径为 6.7、12.1 和 17.5 nm(即 C₁₈ A–C 的平均孔径)的孔隙所需的压力分别为 14.2、7.9 和 5.5 MPa。
我们对上述结果的解释是:当停泵、压力释放后,那些接触角θ > 90°的高比例水相流动相会从颗粒的孔隙中被挤出。如果存在接触角滞后现象,那么应当采用后退接触角作为相关参数。
保留之所以下降,是因为流动相无法再接触到颗粒的内部表面——而大部分表面积恰恰位于此处。停泵之前,使用高比例水相流动相之所以能够获得保留,完全是因为色谱柱处于压力之下。由于色谱柱出口端接近大气压,流动相可能会被迫从靠近出口端的颗粒孔隙中排出。
这可以解释那些不涉及停泵操作、但也观察到部分保留下降的实验结果。这种效应会随着柱后系统的阻力大小而变化,阻力取决于通向检测器的管路的直径和长度,以及检测器流通池的设计。
McCormick 和 Karger 曾提出相同的机制,用于解释使用纯水流动相时 C₈ 色谱柱上 D₂O 洗脱时间的变化。此外,Fadeev 和 Eroshenko 报道了在烷基键合多孔硅胶上进行的水压汞测量结果,其行为与 Washburn 方程一致。
支持该机制的证据如下:
(i)释放压力会导致保留下降,而向色谱柱施加压力可以恢复保留;
(ii)停泵后死体积的减小量与颗粒内孔容积相当;
(iii)保留下降对孔径的依赖性:孔径越大,下降越小。
在通过施加压力部分恢复 C₁₈ 键合相柱保留的实验中,发现需要 22.7 MPa 或更高的压力才能恢复保留。这与孔径为 6.7 nm、接触角在 110° 左右的计算压力大致吻合。
另一个支持高比例水相流动相被挤出孔网络的证据来自停泵前后色谱柱称重的测量结果。对于低覆盖度的 C₁₈ 键合相以及含有氨基甲酸酯基团的键合相,观察到的保留下降较小,这与这些材料对高比例水相流动相具有较低接触角的预期一致。
尽管这些材料预期具有较低的接触角,但我们尚未找到可靠的方法来测定这些数值。该模型还可以解释为什么将甲醇浓度提高到 40% 可以恢复原始保留。
增加甲醇浓度会降低接触角,当 θ<90∘ 时,流动相能够进入孔网络。如果假定在石蜡上测得的接触角可以作为高表面浓度 C₁₈ 键合相的合理模型,那么需要 40% 甲醇才能润湿该 C₁₈ 材料与 Janczuk 等人的结果是一致的。
该机制还可以解释 Li 等人报道的光学透射行为,因为流动相从孔隙中被挤出会产生固/气界面,从而强烈散射光。
尽管这个简单的描述解释了上述大部分结果,但实际情况更为复杂。首先,颗粒的孔结构并非由不相交的圆柱形孔组成,而这是严格使用 Washburn 方程进行解释时的假设。
在图 5 的计算中,我们进一步简化了分析,假设所有孔径相同。这些颗粒的真实孔结构是由不同尺寸和形状的相互连通的孔隙构成的。Conner 及其同事已经描述了在压汞实验中这类孔网络的行为。
与本工作相关的一点是:非润湿液体侵入孔网络是由孔结构中的狭窄处(喉道)控制的,而挤出则是由网络中的较大开口控制的。这意味着将非润湿液体压入孔网络所需的压力将高于液体从孔中被挤出的压力。这在水压汞研究中已得到证实。
另一个复杂因素来自烷基链的流动性。Nagae 等人已经证明,停泵再启动后的保留下降随链长变化,从 C₃₀ 到 C₁₈ 到 C₈ 保留下降依次增加。此外,对于 C₁₈ 和 C₃₀ 键合相,他们发现保留下降随温度升高而增加。
Bidlingmeyer 和 Broske 对 C₁₈ 键合相也报道了类似的结果。上述机制难以解释这些现象,因为接触角预计不会随链长变化。他们对此结果的解释是:当温度高于烷基链的熔点时,流动相更容易从孔中被排出。温度依赖性可能相当复杂,因为表面张力、接触角和表面结构都随温度而变化。
结论
上述机制有助于预测反相色谱柱在高比例水相流动相下的行为。所有关于使用此类流动相时停泵后再启动导致保留下降的报道均表明:基于较小孔径硅胶(平均孔径 < 20 nm)的高表面浓度、封端烷基键合相会出现严重的保留下降。
而基于较大孔径硅胶的高表面浓度、封端烷基键合相,其保留下降则小得多。然而,由于这些大孔径材料的比表面积较低,它们并非最大化极性分析物保留效果的良好选择。
所有关于含有极性官能团的键合相在停泵再启动后未出现保留下降的报道均表明,在键合相中引入极性基团是防止此类保留下降的有效方法。但其中一些键合相对分析物(尤其是碱性化合物)的保留能力相较于传统烷基键合相有所降低。
最大化极性分析物反相保留效果的另一种解决方案是:在较小孔径(约 9–10 nm)硅胶上使用低表面浓度、封端的 C₁₈ 键合相,例如材料 C₁₈ E。这类键合相不仅在停泵再启动后表现出极小的保留下降,而且对极性分析物具有优异的保留能力和峰形。